Theorie
Simulation von Reaktionen an Festkörperoberflächen durch Dichtefunktionaltheorie (DFT) und kinetische Monte-Carlo-Simulationen (kMC)
|
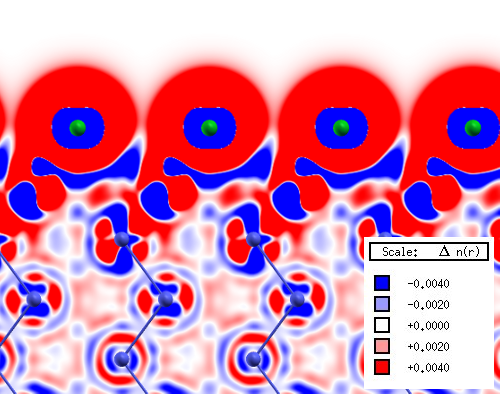
Im Rahmen des Arbeitsschwerpunktes Theorie verwenden wir Dichtefunktionaltheorie (DFT) Methoden zur Beschreibung von Modellkatalysator-Oberflächen, um ein besseres Verständnis der atomaren Prozesse zu erreichen [1]. Dies erleichtert die Analyse und Interpretation der experimentell erhaltenen Daten. Da es sich bei den von uns benutzten Systemen um Festkörperoberflächen handelt, werden periodische DFT -Rechnungen verwendet. Unsere Arbeitsgruppe benutzt das Rechenpaket Vienna Ab initio Simulation Package (VASP) [2]. Mit unseren Rechnungen simulieren wir eine Vielzahl von physikalischen und chemischen Systemeigenschaften. So suchen wir nicht nur nach stabilen Minimumkonfigurationen, z.B. eines Adsorbatsystems, welche wir dann mit experimentellen Strukturdaten z.B. einer LEED-Analyse vergleichen können, sondern modellieren auch die zwischen diesen Minima liegenden Reaktionspfade und deren Aktivierungsenergien um katalytische Reaktionen besser zu verstehen [3, 4]. Ein weiterer wichtiger Faktor , wie theoretische Methoden zum besseren Verständnis eines Problems beitragen können, ist die Visualisierung. Ein gutes Beispiel hierfür sind Dichtedifferenzplots. Diese zeigen die in einem System real vorhandene Elektronendichte abzüglich der Elektronendichten der freien Atome, geben also sehr anschaulich Informationen über Bindungsverhältnisse wider. Die zu berechnenden Details einer Modelloberfläche ergeben sich, je nach Projekt, aus der Art der vorhandenen experimentellen Daten mit denen man sie vergleichen möchte. Wir berechnen hier zum Beispiel harmonische Frequenzen zum Vergleich mit RAIRS Daten (Reflektions-Absorptions-Infrarot-Spektroskopie), Surface Core Level Shifts die eine Hilfestellung für die Auswertung von HRCLS Daten geben und die Simulation von STM Bildern. Ein aktuelles Projekt behandelt die Adsorption von Chlor auf Ru(0001). Aus LEED-Mustern war zunächst bekannt, dass die wahrscheinlichste Minimumstruktur eine (sqrt3 x sqrt3)R30° -Cl-Überstruktur (Bedeckung 1/3) ist. Dies wurde daraufhin mit DFT-Rechnungen überprüft in dem eine Vielzahl theoretisch möglicher Bedeckungen berechnet und energetisch verglichen wurden. Die experimentellen Befunde konnten dabei zweifelsfrei bestätigt und zusätzliche Informationen über das System gewonnen werden.
Rechner:
Kooperationspartner: |
Literatur
|
|
Ansprechpartner