Bioinformatics and Systems Biology
- Overview
-
Today, massively parallel DNA sequencing or hybridization approaches allow the identification of not only the gene repertoire but also the gene regulatory networks of an organism. The huge amounts of data acquired from such experiments can only be handled with intensive bioinformatics support that has to provide an adequate infrastructure for storing and analyzing these data. Thus, bioinformatics has to deliver efficient data analysis algorithms, user-friendly tools and software applications, as well as extensive hardware infrastructure for answering such questions.
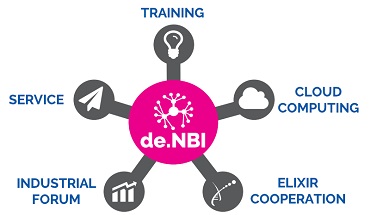
As part of the Bielefeld-Giessen Resource Center for Microbial Bioinformatics (BiGi) , a service unit of the ' German Network for Bioinformatics Infrastructure – de.NBI ', the group is focused on data management for genome and post-genome research projects that require new software solutions for systematic data acquisition, secure data storage of structured information, and high-throughput data analysis. Bioinformatics training and education and the cooperation within the German bioinformatics community is a main scope of the group.
More...
 News
News
 Software
Software
 Publications
Publications
 Research
Research
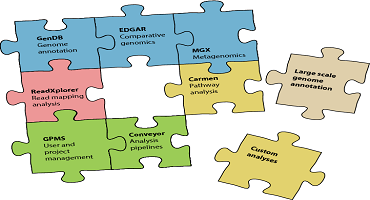
 BiGi
BiGi
 People
People
 Education
Education
 Core Facility
Core Facility

Welcome to the online presence of jlab , short for our working group at Justus Liebig University Giessen: Algorithmic Bioinformatics.
Established in 2019, our core interests are statistical methods for microbiome analyses and combinatorial techniques for sequential data.

Microbiome
The microbiome is the complex community of hundreds to thousands of species from the domains of bacteria, archaea, viruses, fungi and small protists. These microorganisms can be found on all niches on earth, also on and in plants and animals. Actually, the human body should be thought of as a super-organism, since it is composed of roughly the same number of microbial cells as human cells. Utilizing high throughput DNA sequencing, which produces giga- to tera-byte of raw data for hundreds to hundred thousands of microbial samples, we develop tools to process and analyze these gigantic datasets in order to investigate the rich interplay between the microbiome and its host organisms.
![]()
Infrastructure
As partners of the NFDI4Microbiota consortium by the National Research Data Infrastructure Germany, we aim to democratize microbiome studies by providing bioinformatic services and training to scientists in the microbial community. With the lead in the task area services, our goal is the development of a central platform for data deposition and searching for datasets in public repositories, as well as supply analytical services like the microbiome data analysis platform Qiita to a new audience of users.

Algorithm Design
Analyzing sequential data is in many cases based on the algorithmic technique of Dynamic Programming. By developing the language "Bellman's GAP" and an according compiler , we are able to clearly separate independent concerns of Dynamic Programming algorithms which are typically amalgamated in implementations and hamper extending these programs to novel problem formulations.
People
Members of the team "Algorithmic Bioinformatics".
Publications
Work published by members of the team "Algorithmic Bioinformatics".
Teaching
Available Bachelor/Master theses topics.... or bring your own project!
Contact
How to reach us.
Calendar
It might ease scheduling an appointment if you are able to take a look at our availabilities:
(Stefan's core working hours are from 9am to 5pm European Central Time / UTC+01:00)
Profile
One of the BCF's key tasks is the structured acquisition and storage of experimental data and – to the greatest possible extent – the automated processing of that data. It is achieved with the aid of a compute cluster that accesses the required data via high-performance network links on storage and database systems.
Computing Infrastructure
In addition to a flexibly deployable general purpose compute cluster the BCF operates several special purpose computer systems in order to be able to cope with the ever increasing requirements for complex bioinformatics analyses. Within the German Network for Bioinformatics Infrastructure (de.NBI), Justus-Liebig-University Giessen was selected as one of the sites for hosting the de.NBI Cloud - a fully academic cloud, that offers access to large-scale storage and computing resources free of charge for academic users.
Global Access
All services provided by the BCF are accessible from almost everywhere in the world by either web interfaces or directly via remote command line login. Deploying virtualization technology it is possible to allocate specific ressources to individual users which represents a convenient and cost-effective tool for national and international collaboration that often involves the processing of huge data sets.