Bioinformatics & Systems Biology
- Overview
-
Today, massively parallel DNA sequencing or hybridization approaches allow the identification of not only the gene repertoire but also the gene regulatory networks of an organism. The huge amounts of data acquired from such experiments can only be handled with intensive bioinformatics support that has to provide an adequate infrastructure for storing and analyzing these data. Thus, bioinformatics has to deliver efficient data analysis algorithms, user-friendly tools and software applications, as well as extensive hardware infrastructure for answering such questions.
As part of the Bielefeld-Giessen Resource Center for Microbial Bioinformatics (BiGi), a service unit of the 'German Network for Bioinformatics Infrastructure – de.NBI', the group is focused on data management for genome and post-genome research projects that require new software solutions for systematic data acquisition, secure data storage of structured information, and high-throughput data analysis. Bioinformatics training and education and the cooperation within the German bioinformatics community is a main scope of the group.
More...
 News
News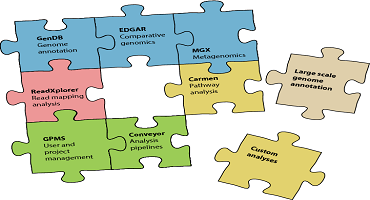 Software
Software Publications
Publications Research
Research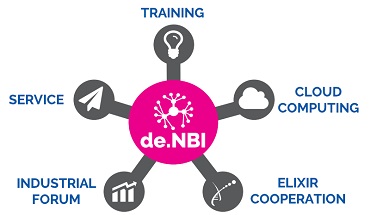 BiGi
BiGi People
People Education
Education Core Facility
Core Facility