Proteomic changes during fibrogenesis
Liver fibrosis is characterized by an imbalance of matrix breakdown, mainly forced by matrix metalloproteinases (MMPs), and matrix synthesis. Tissue inhibitor of metalloproteinases-1(TIMP-1), the key-inhibitor of MMPs, is up-regulated during hepatic fibrogenesis [3, 4]. Hepatic stellate cells (HSC), which are actually discussed to be the main inductor of fibrogenesis, undergo transformation from a quiescent stage into an activated myofibroblast like phenotype. During activation these cells express fibrotic proteins (e.g. fibrillar collagens), TIMP-1, and inflammatory cytokines, maintaining fibrogenesis and extracellular matrix (ECM) accumulation [5].
The rodent CCl4 model resembles the development of toxic liver fibrosis and cirrhosis in men [6, 7]. Abcb4-/- (formerly called Mdr2-/-) mice develop hepatic lesions mimicking primary sclerosing cholangitis [8, 9]. Sclerosing cholangitis in these Abcb4-/- mice is a multistep process. Initially the regurgitation of bile from leaky ducts into the portal tracts leads to periductal inflammation and subsequent periductal fibrogenesis. Atrophy and death of bile duct epithelial cells finally causes obliterative cholangitis with characteristic pattern of up-regulated profibrogenic and down-regulated fibrolytic genes. The Abcb4-/- mouse is thus an attractive model to test potential antifibrotic approaches for the treatment of (biliary) liver fibrosis [8, 9].

Sirius red stained liver slices in normal (SRN) and polarized light (SRP) and HE stained livers of healthy and fibrotic mice. In healthy control mice, deposition of distinct collagen fibres was found surrounding central veins and enclosing portal triads representing normal collagen distribution of healthy liver (panels a-b). Mice treated with CCl4 for four weeks displayed a periportal fibrosis characterized by distinct porto-portal septae surrounding the lobules (panels d-e). The histological grade of hepatic fibrosis of mice treated with CCl4 for two times only did not differ compared with naive control mice. Collagen bridges appeared to be broadened and more diffuse in Abcb4 knock out mice (panel g), and diffuse collagen deposition appeared inside the hepatic lobuli along the sinusoids (panel h). Diversely coloured collagen fibres in polarized light micrographs indicate different types of collagen (panels e-h) [24]. Different morphogenic phenotypes may well be an indicator for distinct cellular and molecular pathways of fibrogenesis in these two animal models.
____________________________________________________________________________________
The aim of our study was a systematic comparison of all protein changes during the initial steps and further development of toxically induced hepatic fibrosis and chronic sclerosing cholangitis. We analyzed the proteome changes of two well established mouse models for chronic liver fibrosis, namely the CCl4 model for toxic induced liver fibrosis and the Abcb4 knock out mouse similar to human primary sclerosing cholangitis [6-9]. For the systematic comparison of all hepatic protein changes in these animal models the innovative Difference in Gel Electrophoresis (DIGE) technique, first available in 2002 [10], was used. DIGE allows the separation and comparison of two proteomes in the same gel [11].

DIGE experiment. A representative picture of an overlay of three dye scan-images Cy2, Cy3, and Cy5 is shown. The red spots show a decreased protein expression in CCl4 treated mouse liver whereas green spots show an increase of protein expression compared to control. A: CPS, B: FDH:, C: transferrin, D: SPI, E: Sbp2, F: ALDH1, G: ALDH2, H: GST.
____________________________________________________________________________________
In CCl4 treated animals 40 spots representing significant changes in protein expression were identified. Twelve of these proteins were up-regulated while 28 were down-regulated compared to healthy liver. DIGE analysis of BALB/c-Abcb4-/- mice revealed significant expression changes in 28 proteins (23 proteins up- and 5 proteins down-regulated). Differentially expressed proteins were excised from preparative gels, in-gel digested with trypsin and analysed using MALDI-TOF-MS. Applying PMF analysis, 20 out of 40 differentially expressed proteins were identified in CCl4 treated animals and 8 out of 28 in Abcb4 knock out mice by searching the murine NCBI data base with the Profound algorithm (table 1 and table 2).
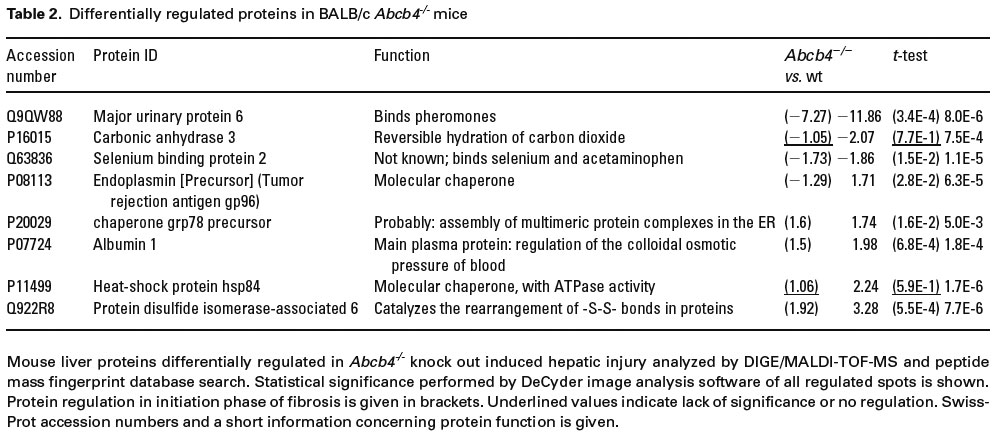{kind=link}
According to DIGE and MALDI-TOF-MS considerable differences in protein regulation during fibrogenesis exist. In contrast to CCl4 induced liver injury, where many of the differentially regulated proteins were down-regulated, up-regulation was found in Abcb4-/- mice (table 1 and table 2). Besides a number of proteins already related to liver injury (e.g. transferrin [26], GSTM1 [27], ALDH1 [28], and ALDH2 [29]) novel proteins have been identified: Sbp2, CPS, SPI, clade A 3K, and FDH. Interestingly, only Sbp2 and carbonic anhydrase 3 – which has already been linked to chronic liver disease [30] - were down-regulated in both models. Thus, the only novel protein associated with liver fibrosis depicting a unidirectional expression pattern in both experimental setups is Sbp2.
Sbp2 was discovered in 1989 [31]. It is mainly expressed in the liver with gender differences in protein abundance [32] and has recently been described in the context of peroxisome proliferator-activated receptor a (PPARa) activation [33]. A chronic PPARa activation leads to a vigorous down-regulation of Sbp2 [33]. In general, Sbps are considered to possess anti-carcinogenic properties and may act like a growth regulatory proteins [34]. In previous studies, Sbp2 was found to bind selenium and acetaminophen for storage or transport purposes but an explicit protein function could not be clarified so far [35].
In the present study, a down-regulation of Sbp2 has been observed in both models of hepatic fibrosis. Previous work considered Sbp2 a selenium transporter, which is predominately incorporated into GSH-Px [36]. Under toxic condition (e.g. alcohol abuse) selenium levels of liver tissue decreased [37]. However, toxic activation and proliferation of HSCs – as seen in CCl4-induced liver fibrosis – may be prevented by selenium supplementation [38]. Subsequently, apoptosis of activated HSCs during acute liver damage may be facilitated [38]. Thus, Sbp2 substitution in toxic induced fibrosis and sclerosing cholangitis may yield a therapeutic strategy targeted against Sbp2 down-regulation.

Protein expression pattern of Sbp2. A: Sbp2 Western blot comparing samples of fibrotic liver (CCl4 8x in 4 weeks; 8 weeks old BALB/c-Abcb4-/-) with the respective control livers. For negative and positive controls the conditioned media of pDE1sp1A (CMV-GFP) and pCMV-SPORT6-mSbp2 transfected COS-7 cells were used. A densitometric analysis of the Western blot is given in Boehringer light units (BLU). B: Differences of Sbp2 mRNA and protein expression levels in the CCl4 model and BALB/c-Abcb4-/- model compared with the appropriate controls.
____________________________________________________________________________________
Just recently a decline in Sbp2 protein content in liver tissue following intraperitoneal paracetamol treatment in mice has been reported [39]. It remains to be elucidated, whether Sbp2 is incorporated in detoxification processes or involved as a transcription factor [39]. Moreover, a rational explanation for the meaning of Sbp2 down-regulation in BALB/c-Abcb4-/- mice is still missing, because oxidative stress does not play a role in the pathogenesis of sclerosing cholangitis. Interestingly, Ahr-/- mice show certain similarities with sclerosing cholangitis in Abcb4-/- mice characterized by periductal inflammation, periductal fibrogenesis and subsequent obliterative cholangitis. Whether Ahr-/- mice exhibit comparable protein expression pattern was well beyond the scope of this study.
The mechanisms leading to CCl4 induced liver injury are not fully understood either, but the initial step seems to involve the bioactivation of CCl4 to the CCl3* radical by cytochrome P450 enzymes [40]. CCl4 toxicity is thought to be initiated by adduct formation of CCl3* with cellular molecules, and formation of the highly reactive trimethylperoxy radical CCl3OO*, which in turn initiates cell destructive chain reactions [41] (i.e. lipid peroxidation, increasing permeability of mitochondrial, endoplasmatic reticulum, and plasma membranes resulting in loss of cellular calcium with subsequent cell damage). However, in this study no CCl3-protein adducts have been detected.
Lipid peroxidation products have referred to as surrogate parameters for oxidative stress. 4-Hydroxy-2,3-(E)-nonenal (4-HNE), a major aldehydic product of lipid peroxidation, is metabolized through oxidative (aldehyde dehydrogenase; ALDH), reductive (alcohol dehydrogenase; ADH), and conjugative (glutathione S-transferase; GST) pathways [42]. The up-regulation of GST, ALDH1, and ALDH2 in toxic liver injury following oxidative stress has been confirmed in our CCl4 model. In contrast, sclerosing cholangitis in BALB/c-Abcb4-/- mice is independent from oxidative stress and consequently GST, ALDH1, and ALDH2 were not detected in the latter model.
In this study two established animal models of hepatic fibrosis were compared. Table 3 summarizes all differentially regulated proteins found via DIGE, for which a direct involvement in liver or fibrotic diseases in general has already been shown.
Limitations and technical advance
Utilizing DIGE we identified proteins that may well be mechanistically linked to hepatic fibrogenesis. The identification of some proteins confirmed the results of earlier studies indicating an association between these proteins and hepatic fibrogenesis (i.e. transferrin [26], GSTM1 [27], ALDH1 [28], and ALDH2 [29]). On the other hand proteomics seems to constitute a powerful tool for identifying new proteins, which have not been related to a specific context before. In the majority of proteins the mRNA expression level revealed a high correlation with DIGE analysis or showed at least the same tendency with regard to either up- or down-regulation.
2D-DIGE technology offers clear advantages compared with conventional 2D gel methods because quantitative analysis and separation of two different samples (e.g. healthy and fibrotic liver) in one 2D gel can be performed. Our group has already demonstrated that DIGE is capable of separating protein lysates from cell lines, primary cells and whole tissue lysates [11]. Beyond this, an internal standard allows the quantification of protein over-expression or down-regulation [50]. In common with traditional 2D electrophoresis DIGE still bears some limitations: specific types of proteins such as membrane proteins and low abundant proteins are underrepresented in 2D gels[11, 51, 52]. Recently it has been shown that CCl4 treatment of mice may affect the mRNA expression of 587 genes (a total of 5180 genes were investigated) [44]. Whether all changes of mRNA expression translate into changes on protein level remains to be demonstrated. Nevertheless, technological advances of DIGE may be capable of identifying more differentially regulated proteins than already found in this study.
In summary, we report a comprehensive comparison of protein changes in two models of liver fibrosis, namely CCl4-induced toxic fibrosis and sclerosing cholangitis in BALB/c-Abcb4-/- mice. Applying DIGE 20 out of 40 differentially expressed proteins were identified in CCl4-treated animals and 8 out of 28 in Abcb4 knock out mice. Obviously relevant differences in the pathogenesis of toxically induced liver fibrosis and sclerosing cholangitis exist: the only novel protein depicting a unidirectional expression pattern in both animal models was Sbp2. However, an explicit protein function could not be clarified so far.
Reference List
[1] Gomez, D. E., Alonso, D. F., Yoshiji, H., Thorgeirsson, U. P. Eur.J.Cell Biol. 1997, 74, 111-122.
[2] Roeb, E., Purucker, E., Breuer, B., Nguyen, H. et al., J.Hepatol. 1997, 27, 535-544.
[3] Murphy, F. R., Issa, R., Zhou, X., Ratnarajah, S. et al., J Biol.Chem. 2002, 277, 11069-11076.
[4] Paquet, K. J., Kamphausen, U. Acta Hepatogastroenterol. 1975, 22, 84-88.
[5] Runyon, B. A., Sugano, S., Kanel, G., Mellencamp, M. A. Gastroenterology 1991, 100, 489-493.
[6] Fickert, P., Fuchsbichler, A., Wagner, M., Zollner, G. et al., Gastroenterology 2004, 127, 261-274.
[7] Popov, Y., Patsenker, E., Fickert, P., Trauner, M. et al., J Hepatol. 2005, 43, 1045-1054.
[8] Zhou, G., Li, H., DeCamp, D., Chen, S. et al., Mol.Cell Proteomics 2002, 1, 117-124.
[9] Henkel, C., Roderfeld, M., Weiskirchen, R., Scheibe, B. et al., Z Gastroenterol 2005, 43, 23-29.
[10] Hiramatsu, S., Kojima, J., Okada, T. T., Inai, S. et al., Acta Hepatogastroenterol. 1976, 23, 177-182.
[11] Siegers, C. P., Bossen, K. H., Younes, M., Mahlke, R. et al., Pharmacol.Res.Commun. 1982, 14, 61-72.
[12] Vidal, F., Toda, R., Gutierrez, C., Broch, M. et al., Alcohol 1998, 15, 3-8.
[13] Lee, H. C., Lee, H. S., Jung, S. H., Yi, S. Y. et al., J.Korean Med.Sci. 2001, 16, 745-750.
[14] Elchuri, S., Oberley, T. D., Qi, W., Eisenstein, R. S. et al., Oncogene 2005, 24, 367-380.
[15] Bansal, M. P., Oborn, C. J., Danielson, K. G., Medina, D. Carcinogenesis 1989, 10, 541-546.
[16] Mattow, J., Demuth, I., Haeselbarth, G., Jungblut, P. R. et al., Electrophoresis 2006, 27, 1683-1691.
[17] Chu, R., Lim, H., Brumfield, L., Liu, H. et al., Mol.Cell Biol. 2004, 24, 6288-6297.
[18] Bansal, M. P., Mukhopadhyay, T., Scott, J., Cook, R. G. et al., Carcinogenesis 1990, 11, 2071-2073.
[19] Ogasawara, Y., Lacourciere, G. M., Ishii, K., Stadtman, T. C. Proc.Natl.Acad.Sci.U.S.A 2005, 102, 1012-1016.
[20] Yang, J. G., Morrison-Plummer, J., Burk, R. F. J.Biol.Chem. 1987, 262, 13372-13375.
[21] Bergheim, I., Parlesak, A., Dierks, C., Bode, J. C. et al., Eur.J.Clin.Nutr. 2003, 57, 431-438.
[22] Shen, X. H., Cheng, W. F., Li, X. H., Sun, J. Q. et al., World J.Gastroenterol 2005, 11, 4957-4961.
[23] Fountoulakis, M., Berndt, P., Boelsterli, U. A., Crameri, F. et al., Electrophoresis 2000, 21, 2148-2161.
[24] Castillo, T., Koop, D. R., Kamimura, S., Triadafilopoulos, G. et al., Hepatology 1992, 16, 992-996.
[25] Weber, L. W., Boll, M., Stampfl, A. Crit Rev.Toxicol. 2003, 33, 105-136.
[26] Luckey, S. W., Petersen, D. R. Arch.Biochem.Biophys. 2001, 389, 77-83.
[27] Lilley, K. S., Friedman, D. B. Expert.Rev.Proteomics 2004, 1, 401-409.
[28] Fountoulakis, M., Juranville, J. F., Tsangaris, G., Suter, L. Amino Acids 2004, 26, 27-36.
[29] Kashino, Y. J.Chromatogr.B Analyt.Technol.Biomed.Life Sci. 2003, 797, 191-216.
[30] Chung, H., Hong, D. P., Jung, J. Y., Kim, H. J. et al., Toxicol.Appl.Pharmacol. 2005, 206, 27-42.